@lakesea
2023-02-27T07:26:37.000000Z
字数 3435
阅读 813
VAG 更新与中文文档 for v1.01
VAG
基于VAG的图形泛基因组reads比对的可视化与审阅,(不止read比对的可视化哈)
VAG是一个图形化泛基因组reads比对和路径导航的可视化程序,根据近一个月的公测的结果,大家反馈了许多问题(bug),我们对此进行了修正,使整体软件效果和效率得到了非常大的提升。本文档旨在提供充分的操作说明,方便研究者进一步使用VAG于科研和生产中。
VAG所支持的图形泛基因组可通过minigraph、cactus等工具进行构建,构建工具生成的GFA文件(需要有特定的参考基因组主线;部分情况下需排序至S行与对应P行交错)需进一步通过gfatools转为Fasta格式文件。
Window下exe版本
随着VAG进一步的成熟,我们为了进一步扩大用户的应用平台,推出了window版本的VAG,且界面进一步优化,目前已经能基本实现从图文件提取,bam文件提取,到可视化展示(目前的版本只支持read比对的结果展示(read)与图形基因组(Graph)展示)。Window版本已整合所有依赖的包,点击即用,通过生成本地图文件与调用浏览器展示可交互的图像。但<.info>文件的生成与泛基因组图的格式的转化仍需通过getinf.py脚本与经gfatools的处理的脚本生成。
对于可视化过程,用户可自由选择面板上的参数后,通过点击Run the visuallization按钮选择由graphsamtools
生成的区间提取文件夹(index)即可进行可视化。在当前文件默认生成PNG图片与调用用户本地默认浏览器打开交互格式可视化图像。
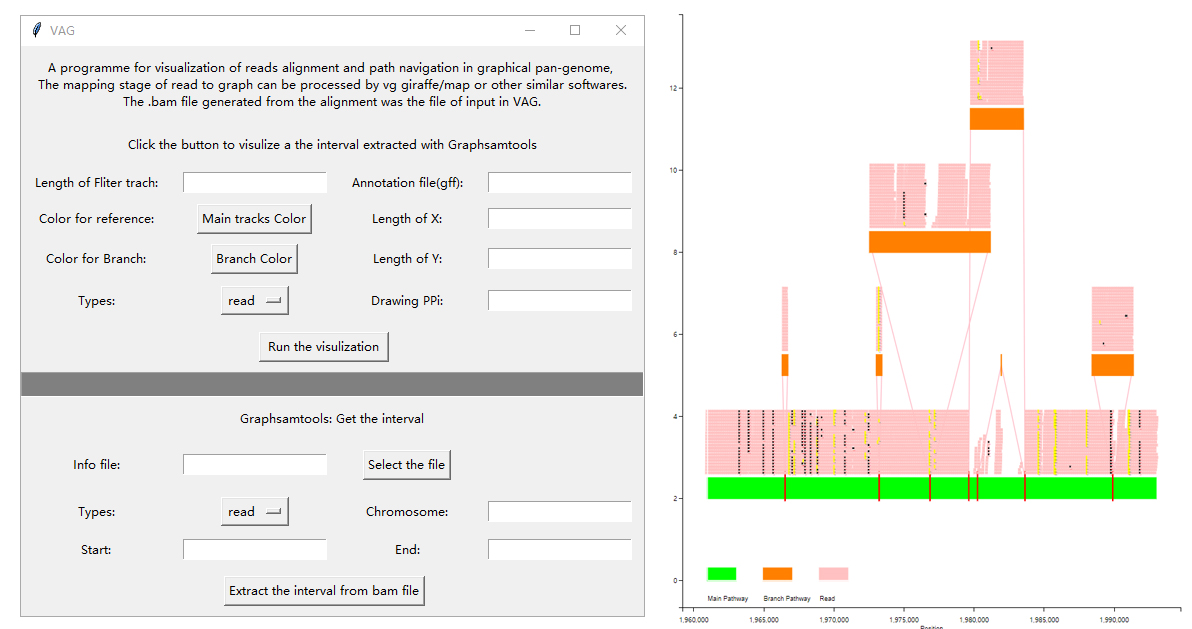
对于序列提取的功能(graphsamtools),由于文件大小与计算过程的所消耗计算资源对于一般window平台机器都是较为吃力的(至少需要40G内存),因此仍推荐在Linux平台上运行。
VAG命令行版本(本体)
以下"<>"内为可变文件命名内容
gfatools gfa2fa -s <图文件名>.GFA > <图文件名>.fa
运用内置脚本getinf.py可将Fasta格式文件转换为VAG进一步所需的图信息文件(info file)
python script/getinf.py <图文件>.fa > <info.文件>
进一步的进入VAG的工作流程。具体可以分为利用graphsamtools脚本基于选定的参考基因组区间为尺度,提取感兴趣的目标区间,并利用runVAG.py进行可视化生产。该过程在默认参数下输出交互格式的out.html文件与out.png文件,这一个部分根据任务分类可以分为:
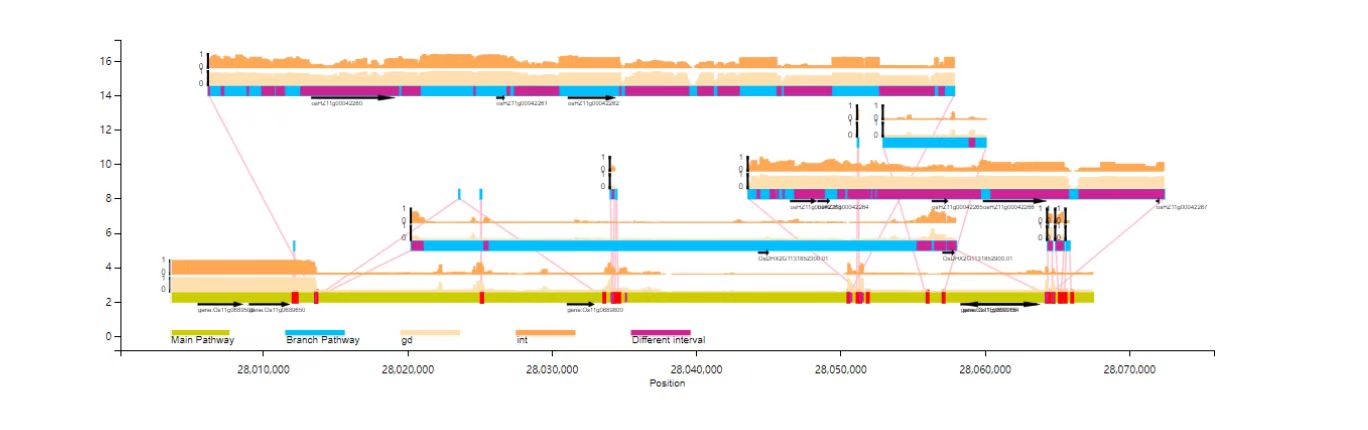
1. 图形泛基因组显示:
graphsamtools <info.文件> <染色体号> <提取起始位置> <提取终止位置> onlytrack <输出文件夹>python runVAG.py --inindex <graphsamtools的输出文件夹> --drawtype onlytrack
效果如下:
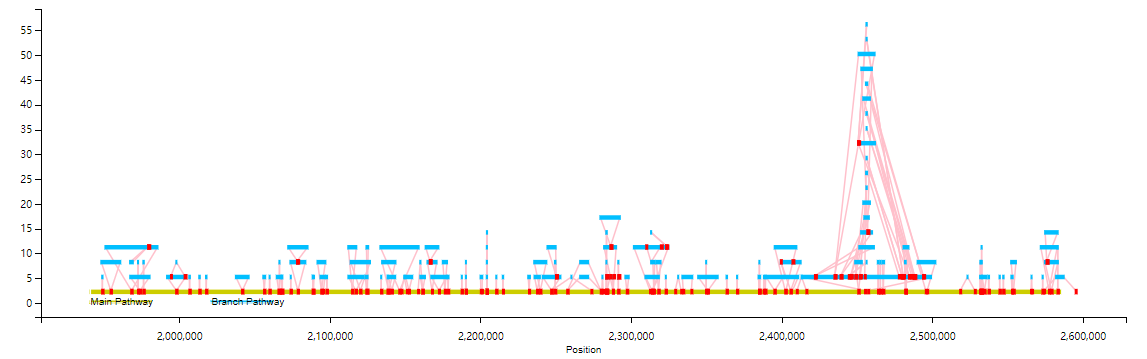
也可通过分支路径的长度进行显示筛选(--fl选项)
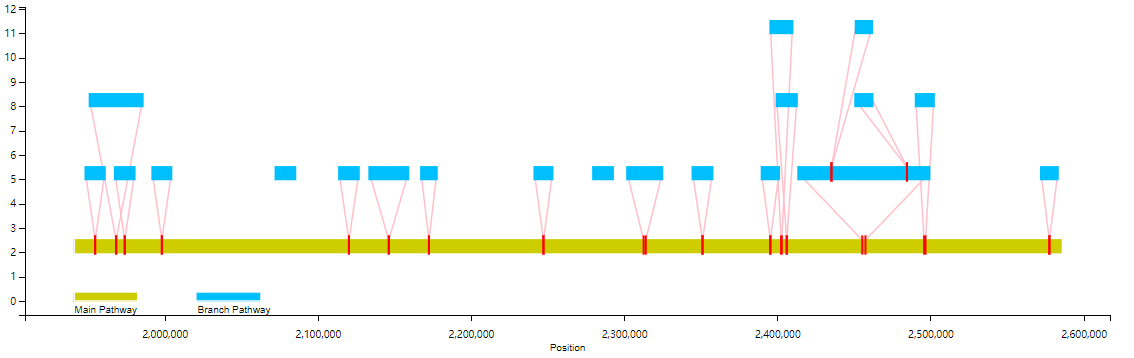
python runVAG.py --inindex <graphsamtools的输出文件夹> --drawtype onlytrack --fl <筛选长度>
比如以下为不显示10000bp以下的分支的效果(branch):
2. 基于图形泛基因组的序列比对(alignment/mapping)的显示:
Reads到graph的映射阶段可以用vg giraffe/map或其他类似的软件来处理。对中生成的.bam文件为VAG的输入文件
graphsamtools <info.文件> <染色体号> <提取起始位置> <提取终止位置> <bam文件> <输出文件夹>python runVAG.py --inindex <graphsamtools的输出文件夹>
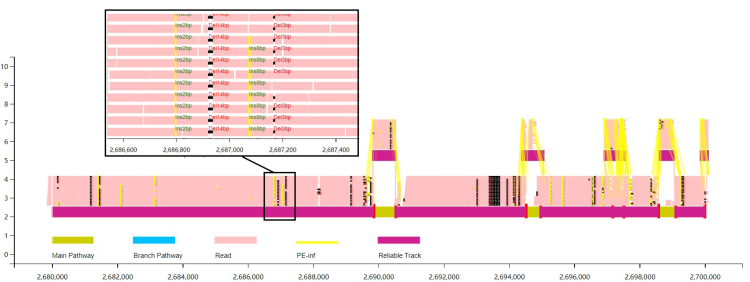
基础效果如下,默认下只显示Insertion/Deletion, mapping信息只显示到深度为20x, 不显示SNP。

可是选择显示短序列比对中的pair-end信息,根据此信息可用VAG进行可靠比对路径的推测。
python runVAG.py --inindex <graphsamtools的输出文件夹> --pairend 1 --anntracks 1
用户可选择将比对的信息的显示为coverage信息(基于mosdepth计算)
graphsamtools <info.文件> <染色体号> <提取起始位置> <提取终止位置> <bam文件> <输出文件夹>python runVAG.py --inindex <graphsamtools的输出文件夹> --drawtype coverage
基础效果如下(柱状图;折线图可选):
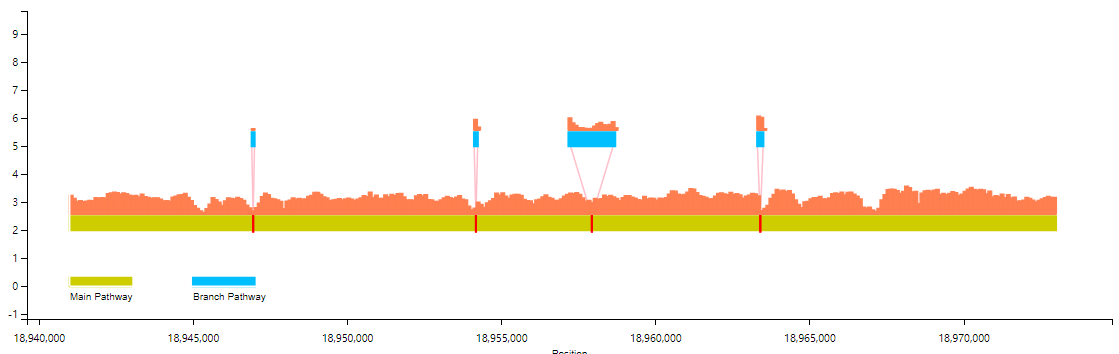
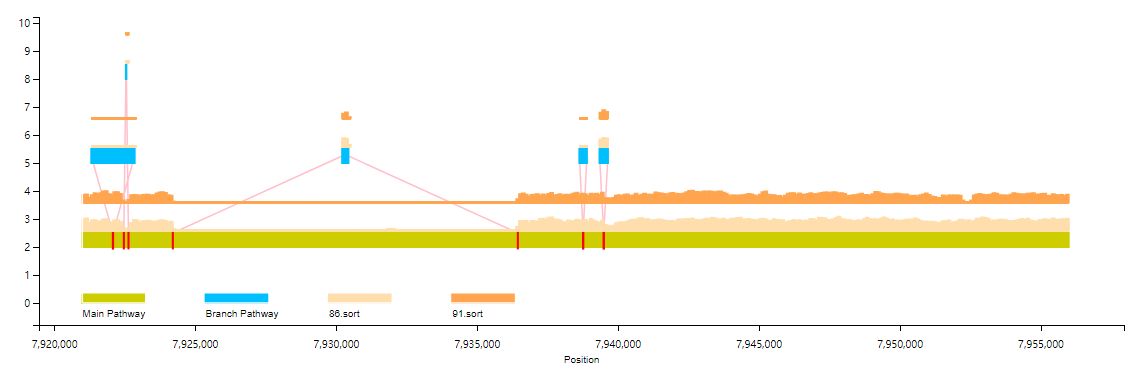
用户也可以选择对多个样品在同一个尺度下进行coverage信息展示(三个样品以上只支持折线图展示)
graphsamtools <info.文件> <染色体号> <提取起始位置> <提取终止位置> mutiplesamples <输出文件夹> <bam文件列表>python VAG/runVAG.py --inindex <graphsamtools的输出文件夹> --drawtype mutiplesamples
#bam文件列表文件示例 (不包含此行):sample1.bamsample2.bam
3. 不同群体间的Coveages frequency (PAV) 也可通过类似的方式进行展示
输入文件为将群体PAV frequency按plink --maf功能处理后的.freq文件(目前只支持两个群体; 默认下标识经过的卡方测验(P<0.01)的差异区域)
graphsamtools <info.文件> <染色体号> <提取起始位置> <提取终止位置> population <群体1.frq>,<群体名称1> <群体2.frq>,<群体名称2> <输出文件夹>python VAG/runVAG.py --inindex <graphsamtools的输出文件夹> --drawtype populationfreq
柱状图:
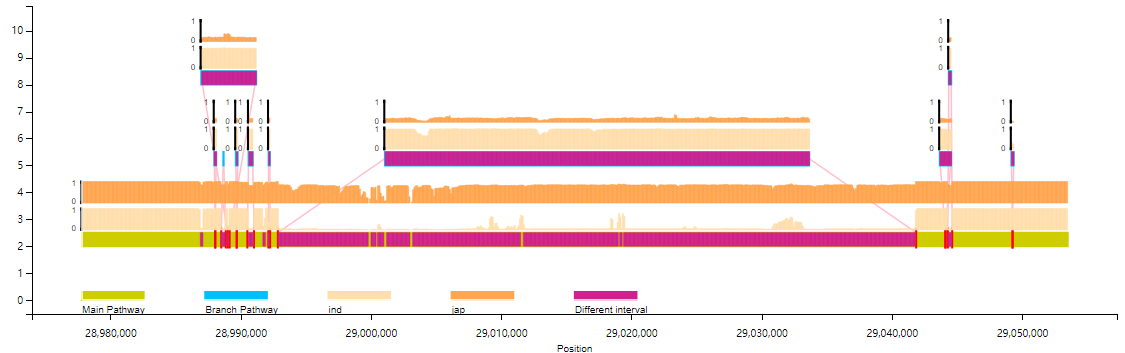
python VAG/runVAG.py --inindex <graphsamtools的输出文件夹> --drawtype populationfreq --popline 1
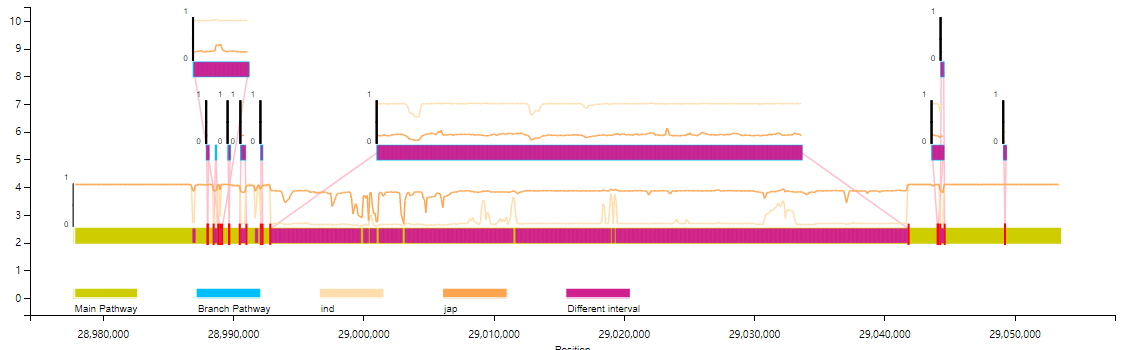
折线图:
python VAG/runVAG.py --inindex <graphsamtools的输出文件夹> --drawtype populationfreq --popline 1
#群体frq文件示例(不包含此行):CHR SNP A1 A2 MAF NCHROBS1 Chr1_950 A G 0.3237 4141 Chr1_1050 A G 0.04348 4141 Chr1_1150 A G 0 4141 Chr1_1250 A G 0 4141 Chr1_1350 A G 0 4141 Chr1_1450 A G 0 4141 Chr1_1550 A G 0 4141 Chr1_1650 A G 0 4141 Chr1_1750 A G 0 414
4. 注释文件的添加
基因/TE等位点注释文件可添加入VAG中联合可视化:
python VAG/runVAG.py --inindex <graphsamtools的输出文件夹> --drawtype populationfreq --gaingene 1 --gff <gff格式的注释文件>
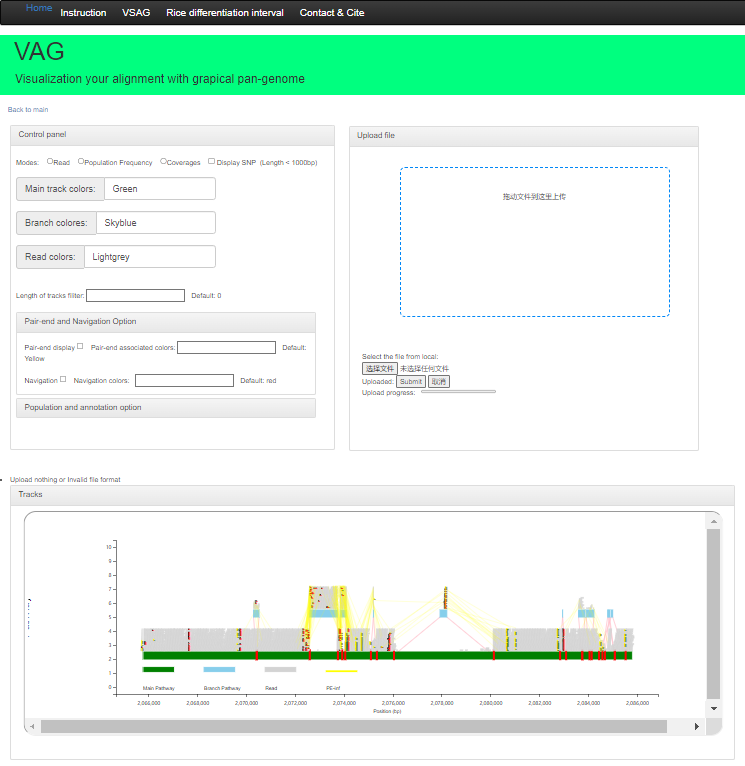
Web版本
Web版本支持基于Apache框架的部署,依赖于php环境。用户可通过对压缩后上传压缩包进行可视化。
tar zcvf <graphsamtools的输出文件夹>.tar.gz <graphsamtools的输出文件夹>
如果您在使用中有任何问题或意见,欢迎与作者联系(lpf_bio@foxmali.com/fangping.li@scau.edu.cn),我们会及时回复!
该工作由华南农业大学联合广东省农业科学院水稻所分子育种团队完成,如果您的研究使用了我们的工具,请引用论文: Visualization and review of reads alignment on the graphical pan-genome with VAG Fangping Li, Haifei Hu, Zitong Xiao, Jingming Wang, Jieying Liu, Deshu Zhao, Yu Fu, Yijun Wang, Xue Yuan, Suhong Bu, Xiaofan Zhou, Junliang Zhao, Shaokui Wang bioRxiv 2023.01.20.524849; doi: https://doi.org/10.1101/2023.01.20.524849,
