@lakesea
2018-08-10T02:47:39.000000Z
字数 2272
阅读 2679
群体遗传专题-学习Fst
群体遗传
概念回顾
Fst:群体间遗传分化指数,是种群分化和遗传距离的一种衡量方法,分化指数越大,差异越大。
两个种群之间遗传差异的基本测量是统计量FST。在遗传学中,F一词通常代表“近亲繁殖”,它倾向于减少群体中的遗传变异。遗传变异可以用杂合度来衡量,所以F一般表示群体中杂合性的减少。 FST是与它们所属的总群体相比,亚群体中杂合性的减少量。
具体可以下面的公式表示:
Fst= (Ht-Hs)/ Ht
Hs:亚群体中的平均杂合度
Ht:复合群体中的平均杂合度
理论上计算Fst的步骤
理论上要估算FST,需要以下步骤:
- 找出每个亚群的等位基因频率。
- 查找复合群体的平均等位基因频率
- 计算每个亚群的杂合度(2pq)
- 计算这些亚群杂合度的平均值,这是HS。
- 根据总体等位基因频率计算杂合度,这是HT。
- 最后,计算FST =(HT-HS)/ HT
例子
基因SLC24A5是黑色素表达途径的关键部分,其导致皮肤和毛发色素沉着。与欧洲较轻的皮肤色素密切相关的SNP是rs1426654。 SNP有两个等位基因A和G,其中G与轻度皮肤相关,在犹他州的欧裔美国人中,频率为100%。美洲印第安人与美国印第安人混血儿的SNP在频率上有所不同。墨西哥的样本有38%A和62%G;在波多黎各,频率分别为59%A和41%G,查尔斯顿的非裔美国人样本中有19%A和81%G.这个例子中的FST是什么?
答案如下:其中每个亚群的H值是通过2pq(p和q各代表等为基因的频率)来计算。

生物信息工具计算Fst
当然了,实际中我们并不需要像上述例子那样逐个计算。等位基因频率的信息都藏在snp calling后的vcf中,再使用恰当的工具就可以快速计算出Fst。下面给大家介绍一种比较popular的计算Fst的方法(还有其他方法不限于一种),使用vcftools:
vcftools --vcf test.vcf --weir-fst-pop 1_population.txt --weir-fst-pop 2_population.txt --weir-fst-pop 3_population.txt --out p_1_2_3 --fst-window-size 500000 --fst-window-step 50000# test.vcf是SNP calling 过滤后生成的vcf 文件;# p_1_2_3 生成结果的prefix# 1_population.txt是一个文件包含同一个群体中所有个体,一般每行一个个体。个体名字要和vcf的名字对应。# 2_population.txt 包含了群体二中所有个体。# 3_population.txt 包含了群体三中所有个体。#计算的窗口是500kb,而步长是50kb (根据你的需其可以作出调整)。我们也可以只计算每个点的Fst,去掉参数(--fst-window-size 500000 --fst-window-step 50000)即可。
这个计算过程相当快,然后生成了output file:p_1_2_3.windowed.weir.fst
快速看看生成的文件有什么
CHROM BIN_START BIN_END N_VARIANTS WEIGHTED_FST MEAN_FSTAmTr_v1.0_scaffold00001 1 500000 4543 0.23994 0.182946AmTr_v1.0_scaffold00001 50001 550000 4166 0.222243 0.170822AmTr_v1.0_scaffold00001 100001 600000 4220 0.195063 0.152002AmTr_v1.0_scaffold00001 150001 650000 4478 0.187311 0.151101AmTr_v1.0_scaffold00001 200001 700000 4901 0.190478 0.156037AmTr_v1.0_scaffold00001 250001 750000 4777 0.182011 0.143682AmTr_v1.0_scaffold00001 300001 800000 4222 0.195812 0.15571#第一列:染色体#第二列:计算开始位置#第三列:计算结束位置#第四列:计算区域内变异的数量#第五列和第六列:加权FST和平均FST
结果的出来后,最终要的还是将其转换成图片的形式,废话不多说,上代码:
library(ggplot2)data<-read.table("/home/ricky/Desktop/ws/Amborella/Phylogency/p_1_2_3.windowed.weir.fst",header=T)sc3 = subset(data,CHROM=="AmTr_v1.0_scaffold00003")#这里选取了scaffold 3来观察p <- ggplot(sc3,aes(x=BIN_END/1000000,y=WEIGHTED_FST)) + geom_point(size=0.5, colour="blue") + xlab("Physical distance (Mb)")+ ylab("Fst") + ylim(-1,1)p + theme_bw()
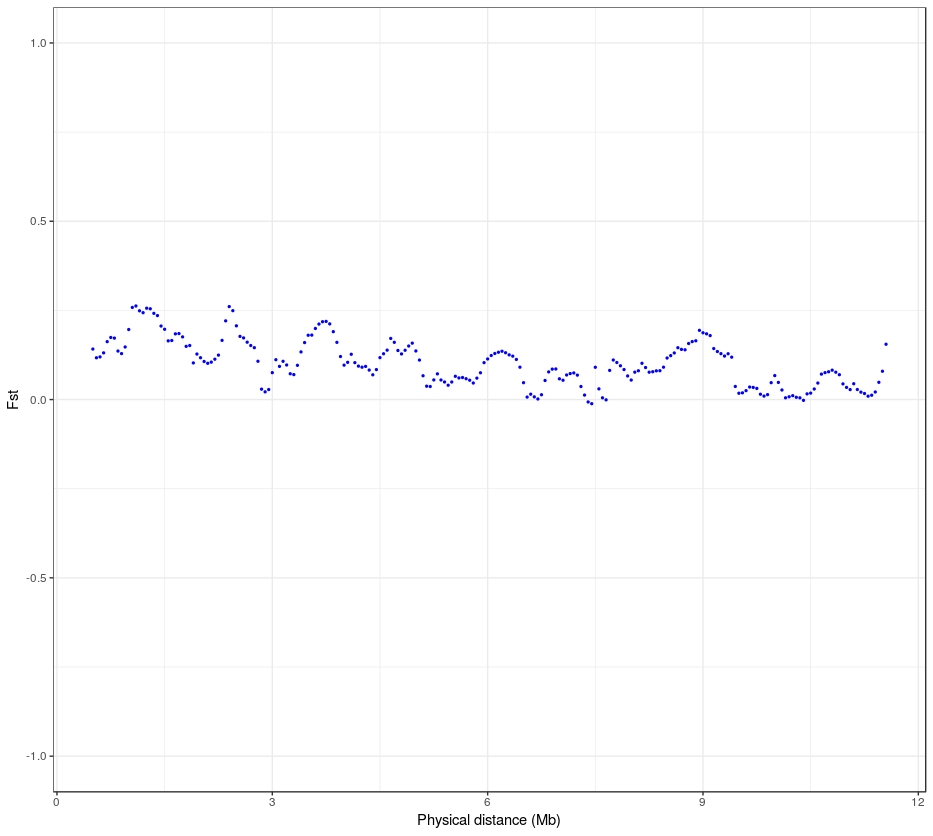
简单解析,在第三个scaffold上没有特别明显的特别大的Fst,在该scaffold上没有明显的驯化分化信号。
理解与应用
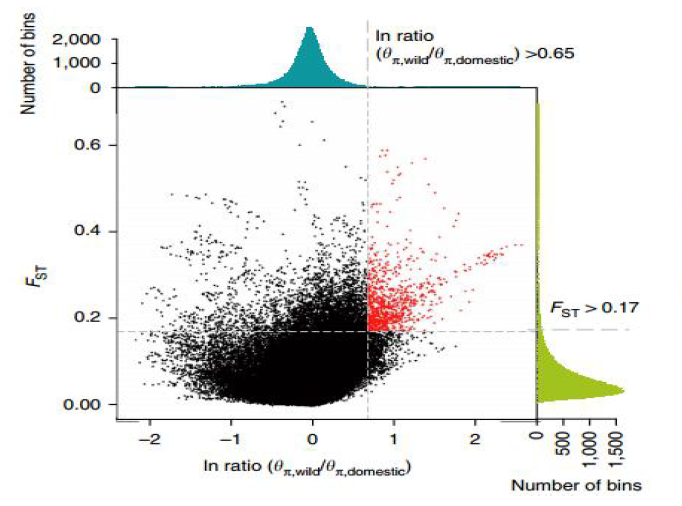
具体的深入应用我还需要多学习一下,但一般可以结合Fst 与 pi 值进行选择性分析,筛选出你感兴趣的位点(如迁移、突变和遗传漂变),下图就是一个列子:通过Fst和pi帅选驯化相关的区域: