@Billy-The-Crescent
2019-07-09T14:14:14.000000Z
字数 10346
阅读 1198
生物信息学
生物信息学 计算生物学 Bioinformatics
目录:
生物信息学引论:
生物信息学期刊:
Bioinformatics
BMC Bioinformatics
Pros Computational Biology
What?
- 在计算机上做生命科学研究的学问
Why?
- 生物学数据越来越多,生物大分子的精细观测数据
从局部到总体,从琐碎的数据得到总体的结论
统计物理学
研究解决某一个问题的方法
- 序列比较
- 相似性比较
- 结构预测
应用已经发展的方法解决生物学问题
- 找同源序列
- 分析进化关系
- 寻找蛋白抑制剂
同义突变对生命体也是有影响的,包括蛋白质的表达强度和蛋白质折叠的情况
研究内容分析:
- 分子数据的收集与管理
数据库搜索及序列比较
Alignment 双序列比对和多序列比对
- 基因组序列分析
人的基因组只有2%的编码序列,如何识别、注释基因,基因调控信息分析,基因组比较 高通量测序技术的应用
全基因组测序、目标区域捕获测序、转录组测序、表观遗传、microRNA分析、宏基因组学
- 转录组学分析
- 蛋白组学分析 质谱的数据,从荷质比推断蛋白质片段。(PEEK)
- 蛋白质结构预测
辅助药物研发
Ligand and receptor docking
- 数据整合与系统生物学
生物信息学常用方法和技术:
- 数学方法
- 运筹学
- 人工智能
- 分子模拟
- 计算机技术
生物数据库资源
数据库 database:
- 有一批数据构成的有序集合,以一定关系储存在一些结构化的数据表中
三类生物数据库:
Swiss-Prot是Uniprot中经过人工矫正过的质量最高的
蛋白质的平均长度是350+
SWISS-Prot有500w+的序列数量,而Uniprot有>1000w个序列
PDB统计:自2008年以来,没有新的蛋白质折叠模式被发现,也就是说,现在所有新发现的蛋白质和已知的蛋白质都是相似的。
序列分析
序列比对
序列决定结构。序列相似性和结构相似性有一定的联系
- 核苷酸比对
- 蛋白质比对
蛋白比对比核苷酸对比更可取,因为ATCG的序列随机产生概率比20种氨基酸的随机序列产生概率要高得多。
比对方法就是一个最大匹配问题
评估不同氨基酸的相似性
如何序列-序列配对
氨基酸比对
如何衡量氨基酸差异:
(利用统计学上、进化上的突变的难易程度来评估两个氨基酸的相似性,突变越容易,那么说明两个氨基酸越相似,这样的比较考虑了进化上的复杂性,更加具有生物学意义)。
相比于以结构和化学性质为基础的比较方式,基于统计学的比较方式虽然更难理解,但是更具有生物学意义。
- PAM矩阵(Point Accepted Mutation)
- BLOSUM矩阵(Block Substitution Matrix)
- PAM矩阵
- 如果两种氨基酸替换频繁,说明自然界接受这种替换,那么这对氨基酸替换得分就高。一个PAM就是一个进化的变异单位, 即1%的氨基酸改变,但这并不意味100次PAM后,每个氨基酸都发生变化,因为其中一些位置可能会经过多次突变,甚至可能会变回到原来的氨基酸。
人和黑猩猩同源蛋白比对时,属近缘关系,选择PAM1;人和酵母菌蛋白比对时,属远缘关系,可选择PAM250
- BLOSUM矩阵
- Blosum直接从最同源的序列的区间排比获取匹配率,不考虑进化距,即BLOSUM62表示的是从同源程度为62%左右的蛋白质家族得到的打分矩阵。
PAM矩阵缺点:一旦PAM1的矩阵有误差,那么自乘250后(PAM250)得到的矩阵的误差就会很大。
(PAM100的100表示自乘次数)
(BLOSUM90的90表示选择的蛋白的平均相似度)
BLOSUM矩阵优缺点:优点是符合实际观测结果,因为衡量了在某一个同源程度上氨基酸的相似程度,缺点是不能提供进化信息。
PAMn是计算出来,而BLOSUMn是观测出来的
双序列比对
如何获得最大匹配?
序列联配(alignment)
- 动态规划:
- 把多阶段过程转化为一系列单阶段问题,利用各阶段之间的关系,逐个求解。一个问题只跟有限个问题有关,因此可以通过那
用表格/数组中计算出来的值而不是用递归重复计算,从而实现一种减少程序负责度的一种方法。(空间换时间)
- 全局比对:
- 寻找序列在全长范围内最佳比对。常用算法为NW算法。
- 局部比对:
- 寻找序列在局部区域的最高比对打分。常用算法为smith-Waterman算法、FASTA算法和BLAST算法。
Needleman-Wunsch算法:
利用动态规划思想,分解序列比对问题。两个氨基酸A,B之间的匹配,无非三种情况:A和B配上了;A和B前面的gap配上了;A和B后面的gap配对上了。
A A - - A
|
B - B B -
NW比对方法:
其中,是空位罚分,在这里定义为-8。表示位置i和j的匹配得分,表示i和k的匹配在氨基酸打分矩阵中的得分。
在NW算法表中,向右意味着上面的那个氨基酸对应空格;向下意味着左边的那个氨基酸对应空格。
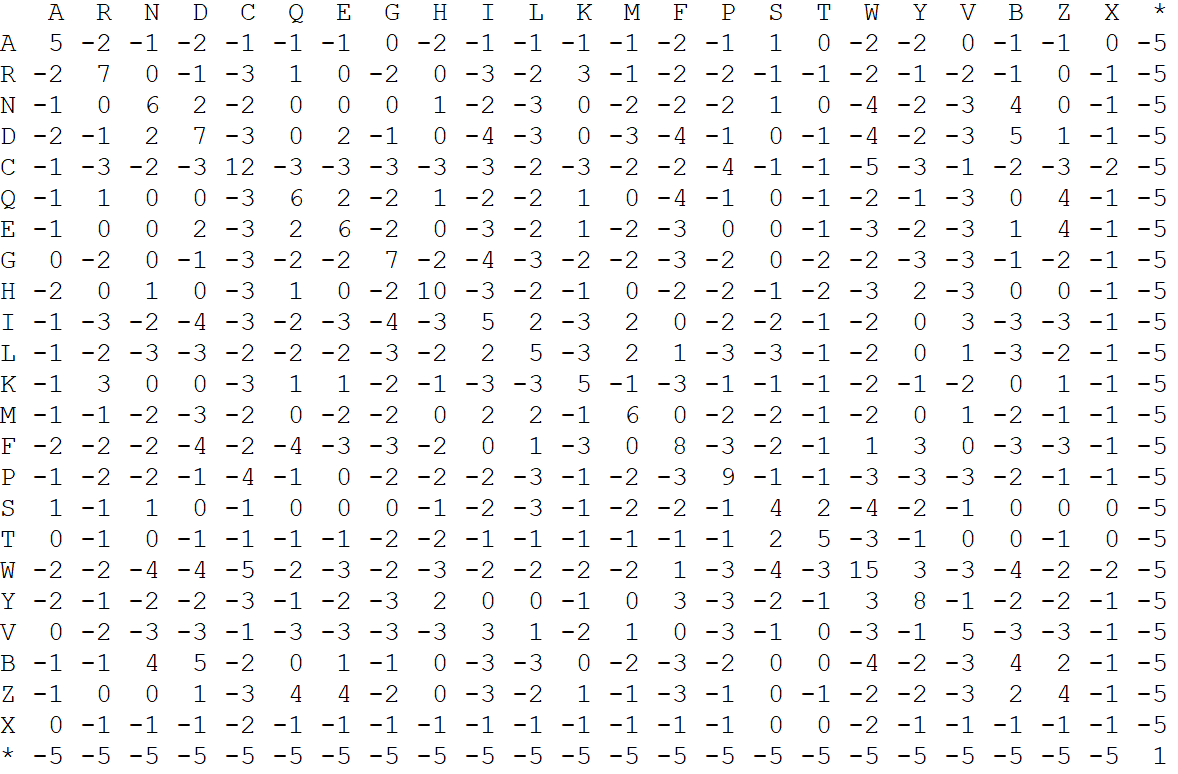
Result:
- 比对的结果不是唯一的
- 循环排列(Circular Permutation)的比对:
用以解决多结构蛋白中 结构域交换的情况 的比对
如:A-B两个结构域蛋白和B-A两个结构域蛋白的比对
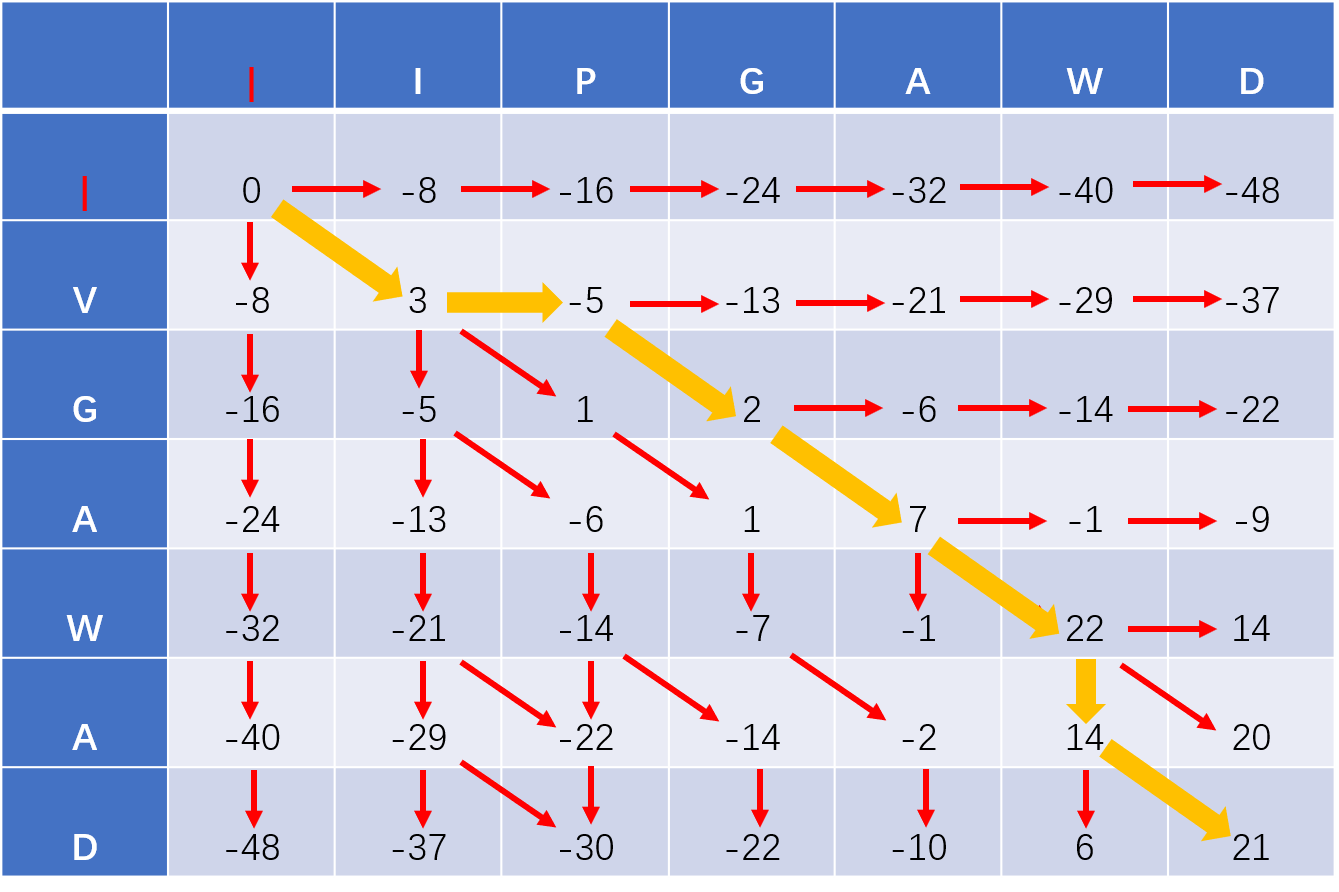
Smith-Waterman算法(只关心高度匹配的区域):
局部配对(局部最大匹配):
回溯的时候,从得分最高的单元格开始,回溯到得分为 0 的单元格为止。
其他优化算法
FASTA算法:一种局部比对算法,使用多个氨基酸的word而不是单个aa进行计算;避开表格边缘的计算,而主要集中在表格对角线上的计算。
事先把配对比较好的配对存起来
BLAST算法:一种局部比对算法,也是将序列分隔成word(seed sequence)
BLAST算法详解
牺牲了一定的比对准确度,但是却显著提高了比对速度。
- BLASTP是蛋白序列到蛋白库中的一种查询。库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
- BLASTX是核酸序列到蛋白库中的一种查询。先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。
- BLASTN是核酸序列到核酸库中的一种查询。库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。
- tBLASTN是蛋白序列到核酸库中的一种查询。与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。
- tBLASTX是核酸序列到核酸库中的一种查询。此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36种比对阵列。
序列比对工具(Tools):
序列比对结果的解读:
假设A-A得1分,A-B得-1分
AAAA
AAAA
得分4
AAAAAA
AAABAA
得分4
那么两个序列,谁的序列相似性更可靠?
- E-value (期望值):
意义:若查询序列为一条随机序列,对于同样的配对空间,有希望找到E条相似性得分为S的序列。m,n为序列长度。
若查询序列为一条随机序列,对于同样的数据库,有希望相似性得分为S的High Scoring Pair (HSP)片段。是搜索空间的大小,和参数与打分系统有关,一般通过Monte Carlo模拟得出。
E值越小,说明这个结果是随机的可能性越小,意义越明确
E < 0.01 可以认定为同源序列;E值介于1和10之间的序列也是一些值得注意的序列。蛋白质序列比对E值上限的默认设置是10.0,核酸序列比对的E值上限的默认设置是2.0,可以根据自己的需要设置E值上限。
同源性和相似性是有本质区别的。同源性表示两条序列有一个共同的进化祖先。
相似性表示两个序列之间相似性的量度。
不能说90%同源,而是说同源或不同源
- 直系同源 vs 并系同源:
- 直系同源 (Orthology)描述在不同物种中来自于共同祖先的基因。Orthologous基因可能有相同的功能,也可能没有。并系同源 (Paralogy)描述在同一物种内由于基因复制而分离的同源基因。
PSI-BLAST:用于发现关系较远的蛋白质
首先进行传统的BLASTp,得到的结果构建PSSM(位置特异打分矩阵),然后用PSSM去搜索数据库,通过相似序列,为每一个位置分别构建氨基酸打分矩阵(BLOSUM/PAM),然后每一个位置根据其自身的打分矩阵来打分,从而获得序列的最终相似度打分。
每一个位置都构建BLOSUM矩阵需要更大的样本量,没有足够样本量的位置可以通过传统的BLOSUM矩阵填充。
使用PSI-BLAST需要:
- 滤除“低复杂度”区域
- 提高建立PSSM矩阵的阈值(例如0.05->0.005)
- 肉眼检视每一次PSI-BLAST循环,注意每一个结果
- PHI-BLAST(Pattern-Hit Initiated BLAST):
- 利用类似于正则表达式的方式来建立模式序列特征,然后通过寻找模式的方法去进行检索。
多序列比对
目的:找出一组序列中的保守片段,以便进行结构、功能上的分析。多序列比对在阐明一组相关序列的重要生物学模式方面起着相当重要的作用。
- 手工比对方法
- 同步法:给定的所有序列同时进行比对,而不是两两比对或分组进行比对。其基本思想是将一个二维的动态规划矩阵扩展到三维或多维。矩阵的维数反映了参与比对的序列数。这类方法对于计算机的系统资源要求较高,通常是进行少量的较短的序列的比对。
- 步进法
步进法:
a. 先进行两两比对。
b. 构建前导树(通过双序列对比结果评估序列之间的关系)
c. 渐进性比对。按照前导树的顺序进行对比,从关系最近的两条序列开始,不断加入新的序列进行比对。形成空位的原则:once a gap, always a gap (ClustalW)
比对过程中,先对所有的序列进行两两比对并计算它们的相似性分数值,然后根据相似性分数值将它们分成若干组,并在每组之间进行比对,计算相似性分数值。根据相似性分数值继续分组比对,直到得到最终比对结果。比对过程中,相似性程度较高的序列先进行比对,而距离较远的序列添加在后面。
Tools:
ClastalW, ClastalX (Windows),
muscle and online version: 1MB max
Clastal Omega: 4MB max
MAFFT and oneline version: 1MB max
当比对序列的相同程度下降时多序列比对结果的准确度也会急剧下降。当相似度<25%的时候尤为明显。
需要使用生物学先验知识去进行人工矫正
识别远亲序列,总结蛋白家族特点:
构造一个序列的pattern,通过多序列比对结果选择出一条能够代表这个蛋白质家族的一致性序列(若一个位置没有代表氨基酸,就是X;若一致性位点是空位,则标记为g),然后使用这个一致性序列去和目的序列进行比对。
一致性序列的g或x和目的序列残基对上时,打分为0.0;一致性序列的gap和目的序列残基对上时,得分-0.5;一致性序列的g和目的序列的gap对上时,得分0.0;··· (如下)
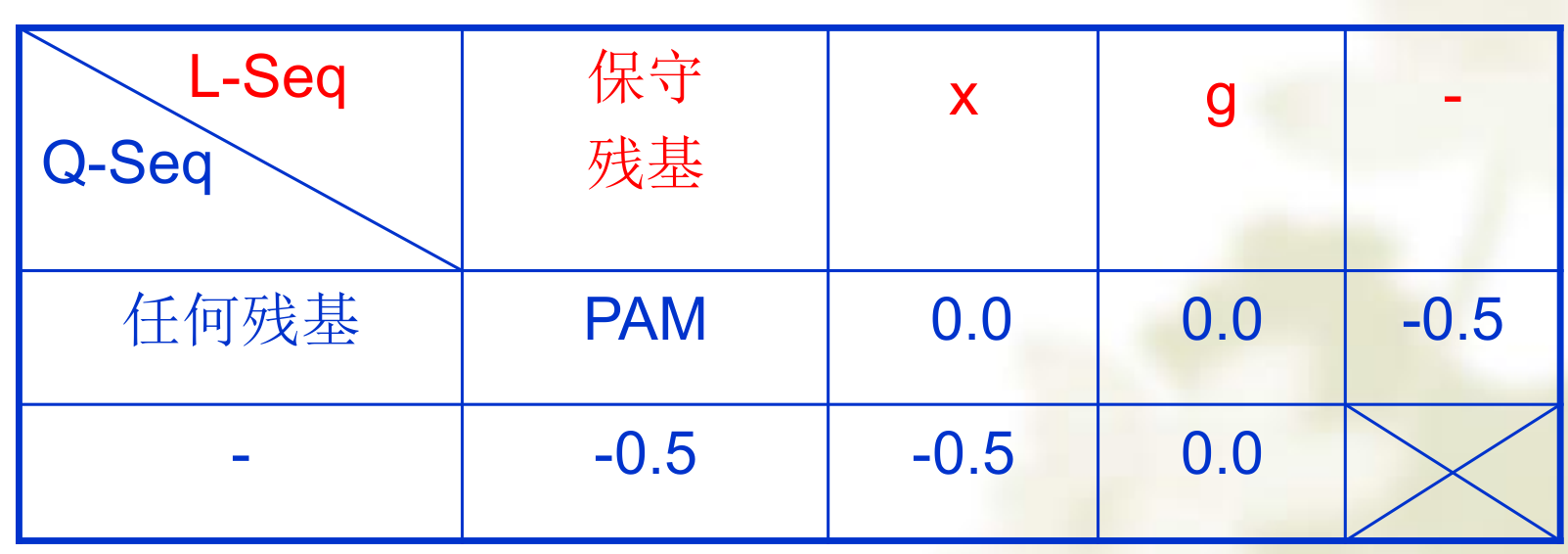
隐马尔科夫模型(HMM)
MPI
HMMER
每一个位点和其前一个位点存在的概率之间的联系,得到每一个位点出现某个氨基酸的概率。得到了一个蛋白的“模式”。
分子进化树
氨基酸位点的“替换”是一个随机过程
中性漂移学说:进化过程中置换绝大部分是中性或近似中性的突变随机固定的结果。
分子钟理论:不同物种同源大分子的进化速率大体相同,如不同物种的血红蛋白分子的进化速度基本上是每个氨基酸每年替换。分子突变的量是可以通过时间积累的。
- 进化树的特征和术语
- 有根树:
- 反映了树上物种或基因的时间顺序。
- 无根树:
- 反映分类单元之间的距离而不涉及谁是谁的祖先。
分子进化树的构建
- 基本思想:
- 物种体内同功能生物分子的相似度越高,则物种的亲缘关系越近。
- 步骤:
- 选择特征分子:各个物种的同源分子;进化速率适当。
对这些分子进行多序列比对,截取比对的最好区域作为物种的代表序列
计算代表序列两两之间的差异度,并进行统计学矫正。
基于这些差异度,绘制系统发生树
对系统发生树进行可行度检验(bootstrap)。
构建系统发生树的方法有:
- 距离法
- 最大简约法 (近源序列)
- 最大似然法 (远源序列)
- 贝叶斯推断
距离法:
1. 对要研究的一组物种,根据某种规则,算出两两之间的距离,构造出距离矩阵。
2. 选出距离最近的两个物种A,C,将其他物种当成一个复合物种。
3. 求出A,C和复合物种三者的分支关系。
4. 从复合物种中取出一个最近的展开,然后将AC复合,求出分支关系。
算法缺点:贪心算法,规定了两个物种之间的相似性,而这不一定准。不能保证找到的是最优的树。
进化树的可信度检验:
- The bootstrap
- Delete-half-jackknifing
- Permuting species within characters
头部的进化数据得到的结果应该和身体部分进化数据得到的结果是一致的,并且通过数据截取、干扰来查看算法的稳定性。
- The bootstrap
- 对“列”进行“有放回地”重取样。从整个序列中任意选取一般,剩下的一般序列随机补齐组成一个新的序列。这样一个序列就变成了多个序列。对每一个序列构建进化树,并进行相互比较以及和原始序列的进化树进行比较,探究每一个分支节点一致的比例。
bootstrap<70时不可靠,bootstrap>=70时一般认为构建的进化树较为可靠。
分子进化的局限性
- 单个分子代表整个物种的片面性问题
- LGT (Lateral Gene Transfer)问题,水平转移
- Gene Duplication使得难以区分orthologous(并系同源)和paralogous(直系同源)的问题
- 方法本身的问题:无法计算回复突变
利用基因组信息构建进化树?
方法:
1. 基本思想:两个基因组之间共有基因的数量之间的差别,构建差异矩阵。将基因组中基因的特征功能划分为17种特征,即两个基因组之间的差别就是这两个17维向量COG之间的差别。
2. 设对应两个染色体(或基因组)的特征矢量为和。用归一化的为和的欧氏距离来代表两个染色体(或基因组)间的距离。
系统发育树常用软件:
PHYLIP
Mega:需要将比对文件转化为.meg格式
- 为什么系统发育树的结果有一个结点没有bootstrap值?
- 因为系统发生树构建的结果只能是无根树,但是显示出来的时候却是有根树,因此有一个结点其实是三分叉,但是为了表示成二叉树,因此有一个结点(根节点)其实是没有bootstrap值。
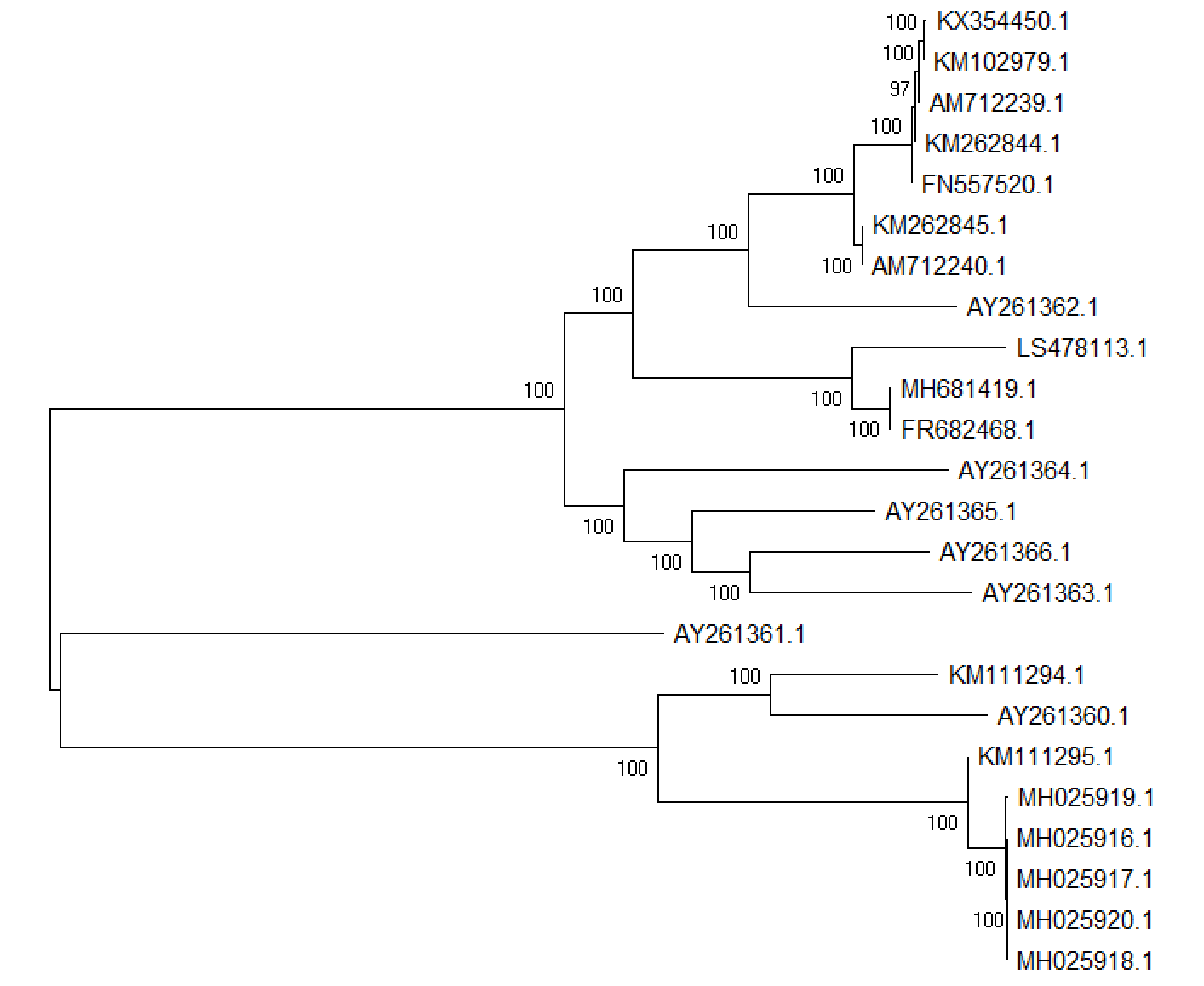
在进行系统发育树分析的时候,一般会人为选择一个物种作为“外群”,以这个“外群”
蛋白质三维结构分析与预测
蛋白质结构
一级结构:
二级结构:alpha螺旋,beta片层
三级结构:单体
四级结构:多聚体
蛋白质结构相似性
用RMSD和TMscore衡量三维结构的差别
RMSD的单位是
最终到一个大小的矩阵
Acta Cryst方法:求解一组向量最大的匹配值
RMSD in ,很相似
RMSD in ,相似
RMSD > , 不相似
软件:TMalign
蛋白质结构预测
序列--结构--功能
历史悠久:超过30年
蛋白质三维结构预测被誉为计算生物学的“圣杯”。
怎样得到折叠好的状态?
能量函数:
- bond length
- bond angle
- dihedral angles
- hydrogen bonds
- van der waals
- electrostatics
搜索算法
- 蒙特卡罗方法
- 遗传算法
- 动态规划
- 宽度/深度优先
- DEE
- 贪婪算法
蛋白质三维结构预测的主要方法
- 同源建模 Homology modeling
- 折叠识别 Fold Recognition/Threading (穿针引线法)
- 从头计算 De novo prediction
同源建模
相似序列具有相似结构
序列相似性低于70%时不适于用在生物学功能解释上。
30%~40%的序列是准确的,但比对上的序列太短也不适合进行同源建模。
- Temperate recognition and initial alignment (模板识别和起始比对):一般使用BLASTn进行比对
- Alignment correction (比对确认):包括确认保守的功能位点以及矫正gaps
- Backbone generation (骨架生成):使用模板的骨架进行建模,保留功能保守位点。
- Loop modeling (循环建模):对于和模板不同源的区域,尝试不同的构象,使得骨架能量最低。
如果有段片段缺失?
利用数学手段,贪婪算法计算怎么将空间上的两个点接起来。 - Side-chain modeling (侧链装配):基于侧链空间构象的rotamer的选择。
- Model optimation (模型优化):自由能最低优化,分子动力学
- Model validation (模型确认):包括功能活性位点是否错误、拉式图等等
ProCheck
WhatIf server - Iteration (迭代):当模型的质量不好时,需要进行迭代运算,直到达到一个比较好的结果。
推荐软件: Modeller,SWISS-MODEL
折叠识别 (穿针引线)
蛋白质的结构近年来越来越多,但是蛋白质的折叠模式却并没有什么变化,即现阶段已经发现了绝大部分的蛋白质折叠模式。
没有单纯利用序列信息,而是利用各种方法(比如二级结构、亲疏水性、PSI BLAST)预测蛋白质的各种属性,利用这些属性进行模板选择,最后执行模型优化和其他的操作。
ITASSER
从头计算
将蛋白质打碎成很多个片段,然后将这些片段进行预测 (自由能最低),最后利用排列组合将这些片段组装在一起,评估哪个组合的自由能最低。
软件:QUARK
分子对接与虚拟筛选
Molecular docking and virtual sifting
预测受体和配体分子形成的复合物结构
分类:
- 蛋白-蛋白分子对接 (口袋不明确)
- 蛋白-小分子的对接 (找口袋 pocket)
蛋白-蛋白对接准确率现在还很低,大约在30%
蛋白-蛋白对接工具:ZDOCK
蛋白-分子对接
重要原则:
- 互补性:决定识别过程的选择性
- 预组织性:决定识别过程的结合能力。受体和配体在结合过程中会有变化。
分子对接减少了水分子的熵,因此是一个能量不利的过程,需要分子对接过程提供一个很强的焓。
搜索算法:如何找到最佳的结合位置
- 遗传算法
- 模拟退火
能量函数:如何评估结合强度
- 基于分子力场的方法
- 基于经验的方法
- 基于知识统计的打分函数
发现结合口袋
- 纯几何学方式
- 从已知数据去发现 (检索同源蛋白已有口袋)
- Lattice Model 格点模型:
- 将蛋白质的结构放在一个有个点组成的空间中,然后随机采样点。以该点为中心往各个方向进行格点扩展,若格点在多个方向扩展到一个长度后通“遇到”了蛋白质内部的格点,那么说明这个地方是凹穴。
- SurfNet:
- 蛋白质空间中随机取采样点,然后扩展成一个球。不断扩展球的半径。如果球的扩展导致球和蛋白各个方向都有重叠的话,说明这是一个凹穴。
缺点:只能发现近乎于球形的凹穴,难以识别扁形的凹穴。
基于几何学方式的方法有缺陷,无法发现形状特殊的口袋。
- 综合信息法:一种集成学习方法
- 综合各种方法的集成方法,将所有方法利用支持向量机 (SVM) 进行学习打分。
方法
COACH:综合信息法
已知口袋坐标进行分子对接
分子对接的分类:
- 刚性对接:研究体系的构想不发生变化。
- 柔性对接:研究体系的构象是可以自由变化的。
利用能量函数计算配体和受体结合前后的自由能变化
分子对接的常用算法:
根据口袋的特征选择配体的构象,进行配体和受体的匹配
| 名称 | 构象搜索方法 | 结合评价方法 | 速度 |
|---|---|---|---|
| Flex X | 片段生成法 | 半经验自由能 | 快 |
| Glide | 系统搜索 | 半经验自由能 | 一般 |
| Gold | 遗传算法 | 半经验自由能 | 快 |
| AutoDock | 遗传算法 | 半经验自由能 | 一般 |
| Dock | 片段生成法 | 分子力场 | 快 |
| ICM-Dock | 随机全局优化 | 半经验自由能 | 快 |
| Fred | 系统搜索 | 半经验自由能 | 快 |
以上方法都不包含口袋搜索步骤。
不需要提前提供口袋信息的方法:
CB-DOCK
分子对接计算的注意点:
小分子问题:
- 起始构象对对接结果有一定影响
- 对分子进行加电荷和加极性氢处理 (需要提供环境pH值)
蛋白质问题:
- 如何选择合理的蛋白质活性位点 (X射线晶体衍射结果蛋白质的N原子和O原子很可能是错的,如果没有出现氢键的话)
- 去除蛋白质中的晶体水分子
对接问题:
- 搜索结合模式的正确性、对接的效率、评分的正确性
- 采用多个软件进行评价,减少结合模式搜索误差
- 定量指标,需要结合分子动力学进一步评价
分子对接未来解决的问题:
- 溶剂化效应
- 分子的柔性
- 打分函数
传统的药物开发方法
乙酰水杨酸 (Aspirin)
- 药物开发从疾病入手
- 从疾病模型寻找药物
虚拟筛选
筛选到的小分子需要进行实验确定效果,然后进行优化。
药物开发的现状:
超过90%的药物不能通过临床试验而被枪毙
平均一款全新药物的研发耗资超过数亿美元,耗时12年。
生物芯片的数据分析
生物芯片种类:
- 基因芯片
- 蛋白芯片
- 组织芯片
- 细胞芯片
差异表达基因的分析
目的:寻找处理前后表达上调或下调的基因
使用T检验,发现显著差异表达的基因。
基因共表达分析
考察基因X和基因Y之间表达的相关性:正相关 or 负相关
方法:
Spearman 顺序相关系数
Paerson coefficient 皮尔逊系数
皮尔逊系数的取值为[-1,1],其中值为+1表示完全正相关,0表示无关,而-1表示完全负相关。
基因表达数据的聚类
K-means聚类
层次聚类
基因表达数据的分类
比如:癌症 vs. 正常组织
与GO数据库关联分析
一个结构化的,旨在建立基因及其产物知识的标准词汇体系,涵盖了基因的细胞组分、分子功能、生物学过程
基因调控网络
利用数学模型建立基因的调控网络。
软件:
GEO-NCBI
Array Express-EMBL
基因组从头测序数据分析
基因组拼装、统计
- 测序基因结果分析
- 基因组定位
- 单核苷酸多态性位点(SNPs)检测和功能分析
- 插入和缺失位点(Indels)检测和功能分析
- 拷贝数变异(Copy Number Variations, CNVs)检测和相关分析
- 其他基因组结构变异(Structural Variation, CVs)检测和相关分析
转录组测序数据分析
有参考基因组 or de novo
基因结构分析
常用软件:
| 功能 | 软件 |
|---|---|
| 识别基因 | GENSCAN,GENOMESCAN |
| CpG岛 | CpGPlot |
| 启动子/转录起始位点 | PromoterScan |
| 转录终止信号 | POLYAH |
| 密码子偏好分析 | CodonW |
| mRNA剪接位点 | NETGENE2,Spidey |
| 选择性剪接 | ASTD |
识别基因:(分原核基因和真核基因)
原核和真核基因的差别:
- 原核细胞无细胞核,基因组呈环状折叠在拟合区域;而真核细胞的基因组高度折叠在染色体中,并和蛋白质高度结合。
- 原核基因没有内含子,且常以单顺反子的形式组织;真核基因大多含有数量众多的内含子,且常以多顺反子的形式组织。
- 原核基因的表观遗传修饰很少,基因暴露;而真核基因一般和组蛋白结合,只在少数区域(转录调控区域)暴露。
- 原核细胞的基因具有较高的一致性,而真核细胞中线粒体、叶绿体的基因和染色体的基因结构有较大的不同,包括在密码子的选择上。
- 原核基因有时存在重叠基因的情况,而真核细胞没有。但是真核细胞有可变剪接机制,一段DNA可以经过转录和转录后加工之后变成多种mRNA。
原核基因基本结构识别
一个核酸序列有六种可能的编码方式,正义链3种,反义链3种。
基因思想:
如果能够找到一个比较长的序列,其相应的密码子序列不含终子密码子,则这个序列可能就是编码序列。(即扫描终止密码子)。然后向前找qishimimaz
判断找到的基因的长度是否过短,若小于64bp的则认为不是基因。
两个软件:GeneMark和Glimmer
真核基因基本结构识别——断裂基因结构
基于同源比对的方式:BLASTn/BLASTx
基于序列特征的方法
序列特征信号:
- 转录启动信号
- 起始密码子
- 外显子、剪接位点 (GT-AG法则)
- 密码子偏好性:包括单密码子偏好性和双联密码子的偏好性
- 转录终止信号 (包含polyA序列)
基因组注释
基因功能分类
比较基因组学及其进化分析
建立数据库
软件:MZEF、GeneScan
